
バイオインフォマティクスとは
バイオインフォマティクスとは
バイオインフォマティクス(英語:bioinformatics)とは、生命科学と情報科学の融合分野のひとつです。広義には生物学、コンピュータサイエンス、情報工学、数学、統計学といった様々な学問分野が組み合わさった学際分野自体を指します。DNAやRNA、タンパク質をはじめとする、生命が持つ様々な「情報」を対象に、情報科学や統計学などのアルゴリズムを用いた方法論やソフトウェアを開発し、それらを用いた分析から生命現象を解き明かしていく、ということが主な目的です。
バイオインフォマティクスの初期の主要な用途はDNAやタンパク質の配列予測でしたが、近年では様々な生体情報の繋がりや相互関係の解析にも用いられています。バイオインフォマティクスが対象とする研究分野は生物学全体に拡大・発展しつつあるのです。
バイオインフォマティクスの例
・膨大なゲノム(遺伝子・DNA)データを解析して病気のメカニズムを明らかにする
・タンパク質の形や動きを詳しく調べ、治療薬を開発する
・生命のネットワークやシステムをコンピューターでシミュレーションする
・ゲノム情報をもとに生命の進化の歴史を解明する
バイオインフォマティクスについて | Japanese Society for Bioinformatics - JSBiから引用
生物学の膨大なデータがコンピューターの演算能力で解析可能に
生物学の進歩に伴い、私たちは生物に関する多くのことを知ることができるようになりました。エンドウ豆の形からメンデルの遺伝の法則が発見された時代には思いもしなかった世界がそこに広がっていたのです。現在、生物学で得られる成果からうかがい知れるのは、膨大な数の物質が複雑に関わり合う世界です。良く研究されているヒトの例では、体を形作る細胞の数は約37兆個、生命の設計図と言われるDNA配列は約30億個、DNA配列の情報をもとに作られるタンパク質は約10万種類あると推定されています。このような世界を捉えるために、「コンピューターの計算能力を活用する」技術が大きな力を発揮し、バイオインフォマティクスと呼ばれる一大分野に発展しています。
バイオインフォマティクスと遺伝子は好相性
バイオインフォマティクスが最も活用されている例として、遺伝子解析を取り上げます。DNAの情報はA, T, C, Gという塩基(RNAはA, U, C, G)で記述できるため、デジタル情報に近い性格を持っています。このことから遺伝子はコンピューターとの親和性が高いと言われています。
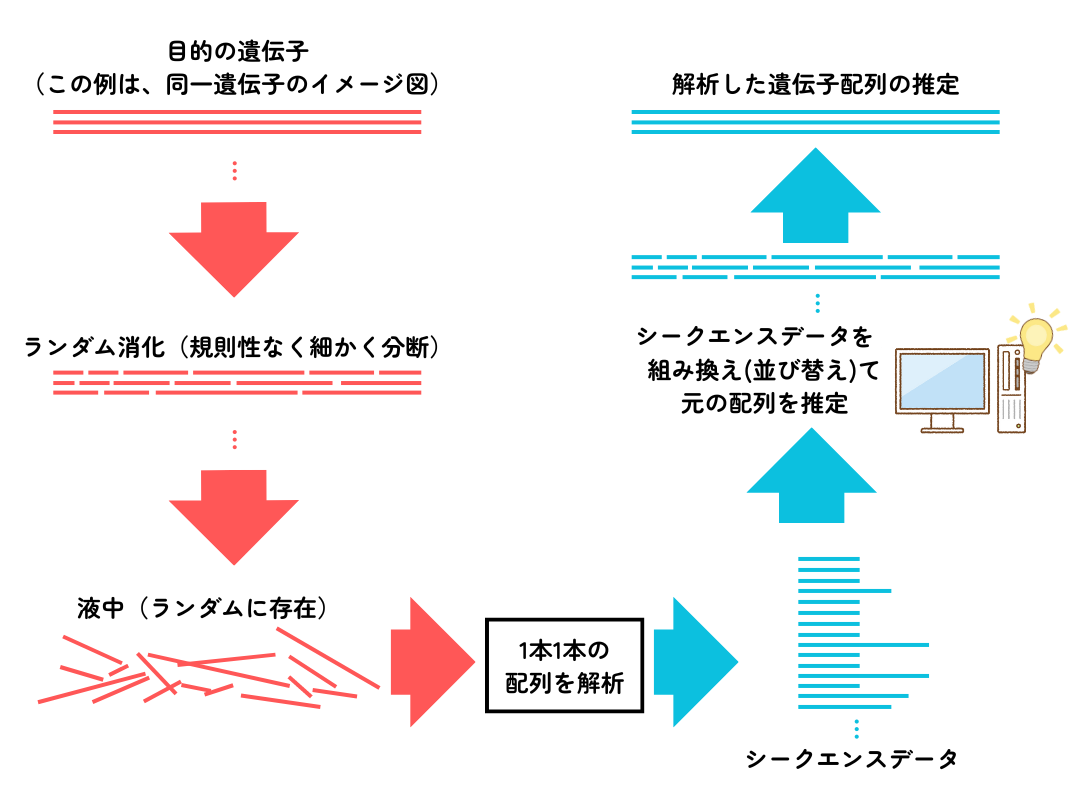
1世代前の代表的な遺伝子解析方法(ダイターミネーター法※1)では、おおよそ1000bpが1回の分析で読み取れる上限とされていましたが、数千万から数十億個の短いDNA断片を1度に解析できる次世代シーケンサー(NGS)という装置の登場により、大幅に読み取り能力が向上しました。NGSでは1回の分析で得られた大量の短い遺伝子読み取りデータを幾重にも重ねてつなぎ合わせることで、元の長い遺伝子配列を推定しています。具体的には、読み取りたい遺伝子を酵素などで消化(細かく分断)し、膨大な数の遺伝子断片それぞれの配列を読み取って、解析ソフトウェアのアルゴリズムで幾重にも重ねてつなぎ合わせて元の遺伝子配列を推定します(下図参照)。
※1 ダイターミネーター法:サンガー法を改良した方法。DNAポリメラーゼの合成反応がターミネーター(ジデオキシヌクレオチド)の取り込みにより停止することを利用した方法。ddATP、ddGTP、ddCTP、ddTTPの4種類を蛍光標識して反応させ、キャピラリー電気泳動を行うことによって泳動時間と蛍光強度により塩基配列を決定する。
一説によると、大規模な遺伝子解析の時代は1987年に自動DNAシーケンサーが世に出たことから始まったと言われています。その後、読み取れる長さと解析速度は急速に進歩し、現在のNGSに至ります。
遺伝子配列の歴史参考:ゲノム研究の歴史と技術革新
これらから得られた遺伝子配列の情報は、データベースとしてWeb上に公開されています。代表的なものとして「EMBL Data Library (現EMBL Nucleotide Sequence Database)(1993年公開)」「Genomes(1995年公開)」「Human Genome(1999年公開)」があります。
世界中の研究者が、研究対象遺伝子のDNA配列と類似するものを検索したり、先行研究を調べたりすることが容易にできるようになり、生物学研究の発展をさらに加速させていきました。
バイオインフォマティクスのはじまりはタンパク質のアミノ酸解析から
バイオインフォマティクスの最初の対象となったのはタンパク質の解析で、その始まりは1960年代初頭までさかのぼります。まだデスクトップ型のコンピューターは存在せず、DNAの配列決定もできなかった時代です。
1959年、フレデリック・サンガー(Frederick Sanger)によって初めてタンパク質のアミノ酸配列がDNP法※2で決定されました。この後、エドマン法で自動解析装置が開発されます。エドマン法※3で配列決定できるのは理論上最大でも50〜60アミノ酸程度でした。そのため、大きなタンパク質配列を知るためには、タンパク質を一度小さな断片にして解析し、得られた配列をタンパク質全体の配列として組み立て直すことが必要でした。この研究者ニーズに応え、コンピューターを利用したアミノ酸配列解析ソフトウェアが開発され、これがバイオインフォマティクスに至る萌芽の一つとなりました。開発したのはマーガレット・デイホフという物理学者です。彼女は後にタンパク質配列データを簡略化することを目的としたアミノ酸の1文字コードも開発しており、(例:アスパラギン=N、アラニン=A、バリン=Vなど65文字から成り、現在も使用されている)、史上初の生物学的配列データベースが完成しました。ゆえにデイホフはバイオインフォマティクスの母と呼ばれています。
※2 DNP法:タンパク質をサンガー試薬(FDNB:1-fluoro-2, 4-dinitrobenzene)で処理するとタンパク質のN末端側のアミノ酸がDNP誘導体として得られることを利用した解析法
※3 エドマン法:タンパク質にエドマン試薬(PTC:phenyl isothiocyanate)を反応させ、N末端のアミノ酸1残基を遊離させてその同定を繰り返す解析方法
広がりを見せるバイオインフォマティクス
タンパク質のアミノ酸配列解析が進み情報量が豊富になるにつれ、その情報は進化学においても活用されるようになります。古遺伝学(Paleogenetics)という新しい学術分野は、生物間で類似したタンパク質のアミノ酸配列の違いからタンパク質の進化の歴史を探ります。タンパク質のアミノ酸配列を類似性が最大になるように整列させて比較(アラインメント)することで、配列の類似性を算出し系統樹をつくるのです。このアルゴリズムは1970年にニードルマンとブンシュ(Needleman–Wunsch)によって開発されましたが、これはバイオインフォマティクスにとって非常に大きな出来事でした(二本の配列を比較するペアワイズアラインメント)。コンピューターアルゴリズムを用いて配列を比較する手法はその後も進化し、BLAST(Basic Local Alignment Search Tool, 1990年)や三本以上の配列を整理するClustalW法(マルチプル・アラインメント, 1997年)などが開発されました。これにより生物学者は、自分が見つけた配列と類似する配列をデータベース上で探し、その遺伝子やタンパク質の機能を推定することが可能となったのです。
DNAの構造解明に伴い、さらに重要性が増すバイオインフォマティクス
ジェームズ・ワトソン(James Watson)とフランシス・クリック(Francis Crick)による1953年のDNAの二重螺旋構造の発表、1958年のクリックによるセントラルドグマの提唱により、タンパク質のアミノ酸配列はDNAを解析することで予測できると考えられるようになりました。1960年代に全アミノ酸に対応したDNAの塩基配列の組み合わせが解明されたことで、DNA配列解読の時代が始まります。DNA配列が分かればタンパク質の一次構造が明らかになるため、世界中の研究者が配列解読に取り組みました。DNA解析技術も進化し、1979年にはサンガー法によって得られたデータをコンピューターで解析するための最初のソフトウェアがロジャー・スターデン(Roger Staden)によって開発されました。
1980年代になると、生物学のデータベースの統合が進みます。1986年に欧州のEMBLと米国のGenBankが、1987年には日本のDDBJ(DNA Data Bank of Japan)が統合されました。(国際塩基配列データベース連携 INSDC:International Nucleotide Sequence Database Collaboration)。このデータベースには、各研究者が解析したゲノムの塩基配列が登録されます。データはすべて無料で公開されており、登録されているDNA配列やアミノ酸配列を検索できるようになったことは生物学の進展を加速させました。
現在も使用されているPerl(1987年)やPython(1991年)といったスクリプト言語もこの時期に登場しています。
ヒトゲノム計画がバイオインフォマティクスの進展の大きな契機
バイオインフォマティクスが大きく発展したのは、1990年から2003年にかけて行われたヒトゲノム計画の時期だと言われています。ヒトゲノム計画は、ヒト細胞の核内にあるDNAの全塩基配列を解読することを目的として、米国政府が国立衛生研究所(NIH)を主体として1990年から15年間で30億ドルの予算を組んで発足した計画でした。当初は15年かかると考えられていたこの計画は、予定より2年も早い2003年に完了しました。プロジェクトの早期完了に貢献したのが、解読速度を大幅に向上させたキャピラリー型DNAシーケンサーと全ゲノムショットガン法です。全ゲノムショットガン法は、大量に得た断片配列を相互の重なりを頼りにコンピューターでつなげ(アセンブリ)、元の配列の長さの10倍程度の長さを読めば大体カバーできるとされています。この方法を開発したのは、ヒトゲノムプロジェクトと競争関係にあったセレラ社という民間企業です。遺伝学者のクレイグ・ベンターが創業しました。
またこの時期にWorld Wide Webが登場し、世界中の生物学データに簡単にアクセスできるようになったことも、バイオインフォマティクスの進展を加速させた大きな要因です。世界初のヌクレオチド配列データベースであるEMBLヌクレオチド配列データライブラリ(SWISS-PROTやREBASEなどの他のいくつかのデータベースを含む)は、1993年にWebで利用可能になりました。1994年には、NCBI BLASTがオンラインで利用可能になりました。さらに「Genomes」(1995年)、「Human Genome」(1999年)といったデータベースが続々と設立され、生物学者は自分の実験データとデータベース上の大量のデータを比較して自分のデータの意味付けを行うことが可能となりました。さらに注目すべきこととしては、データベースに格納されたデータだけで研究するデータ駆動型研究が目指されるようになったことです。仮説をもとに実験し、そのデータをもとに検証する従来の生物学の方法とは全く異なるパラダイムシフトでした。
ヒトゲノム計画の終了後もバイオインフォマティクスの進化は続きます。
2006年に次世代シーケンサーが登場し、生物学のデータは指数関数的に増加する一方、並行して計算科学(コンピューターサイエンス)もその処理能力を大幅に向上させました。
アマゾン、マイクロソフトなどの民間企業によるハイパフォーマンスなコンピューターや専門知識の提供などのサービスも始まっています。
ビッグデータは目的をもって解析し、意味ある解釈を導くことではじめて有用な知見となります。機械学習、人工知能といった新たな技術を活用する試みも始まっており、今後も更なる解析技術の進化が期待されます。
これからのバイオインフォマティクス
2010年代以降にさらに発展している解析分野には以下のようなものがあります。
ゲノムワイド関連解析:
多人数のゲノムを網羅的に解析することで、0.1%ほどとされているDNAの個人差と、疾患のかかりやすさや個々の形質の違いにどのように関連しているのかを統計的に調べようとする試みです。人工知能を組み合わせることで病気の予測に応用する研究も行われ、白血病の診断に役立つという成果も上げています。
タンパク質立体構造解析:
タンパク質の立体構造決定には、X線結晶構造解析や、低温電子顕微鏡、核磁気共鳴などが利用され、多大な時間、労力、予算が必要でした。バイオインフォマティクスによりタンパク質のアミノ酸配列データから立体構造を予測する計算手法の開発が進んでいます。人間のトップ囲碁棋士に勝利したことで話題になったAlphaGoを開発したGoogle傘下のDeepMind社はアミノ酸配列データからタンパク質の立体構造を予測するAlphaFoldという人工知能プログラムを開発しています。AlphaFold2は2020年のタンパク質立体構造予測コンテストで研究者を驚かせる程の予測精度を発揮しています。タンパク質の立体構造は、薬剤の設計において重要な情報であり、予測精度の向上により、医療への応用が期待されます。
遺伝子発現データ解析:
マイクロアレイ技術から得られる大量の遺伝子発現データから、腫瘍細胞を詳細に分類し治療に生かす研究が行われています。さらに、遺伝子の発現量の時間的変化データからは、遺伝子間の制御ネットワークを明らかにする研究も進んでいます。また、次世代シーケンサーを活用して細胞1個ずつの遺伝子発現量を調べるシングルセル解析も始まっています。
生体ネットワーク解析:
生体ネットワークの解析にはゲノムからの転写物の総体であるトランスクリプトームや、トランスクリプトーム(の一部)が翻訳されたタンパク質の総体であるプロテオーム、タンパク質の二次産物として合成される糖鎖の総体であるグライコーム、更にはゲノムからの直接的に転写・翻訳された実体だけではなく、代謝ネットワーク(代謝マップ)によって生じた代謝産物をも含めた総体を考えるメタボローム、生物個体の表現型の総体であるフェノームなどがあります。
またそれぞれのネットワークを独立的に解析するのではなく、統合した生物現象のモデルを構築し、コンピューター上でシミュレーションすることでその妥当性を検証するシステムバイオロジーという研究も取り組まれています。2021年には理化学研究所からマウス脳の全細胞を解析するクラウドシステム“CUBIC-Cloud”が発表されました。
このようなバイオインフォマティクスの発展を支える人材として、バイオインフォマティクスに特化した研究者であるバイオインフォマティシャンの需要は、学術、民間、政府の業界を問わず高まっています。国際計算生物学会(International Society for Computational Biology)はバイオインフォマティシャンを複数のカテゴリに分類し、それぞれが持つべきコアコンピテンシー(技術や能力)を発表しています。
日本国内では1999年に発足した日本バイオインフォマティクス学会が技術者認定試験を主催し、国内の研究室を紹介しています。
(参考)バイオインフォマティクス技術者認定試験の出題範囲
・生命科学分野:
生物学基礎、分子生物学・生化学、バイオテクノロジー
・情報科学分野:
コンピュータシステム、アルゴリズム、データベース技術、確率・統計、機械学習
・バイオインフォマティクス:
分子生物学、配列解析、タンパク質/立体構造・機能解析、進化・遺伝、オーミクス
まとめ
膨大な量のDNA配列を短時間で解読する次世代シーケンサーをはじめ、高性能な解析装置の登場による生物学データの増加と、インターネットの普及によるWeb上のデータベースの発達、コンピューターの計算能力の向上が相まって、バイオインフォマティクスという学問が誕生・発展しました。増加し続ける生物学データを前に、重要性が増している分野です。ビッグデータから新たな知見を生み出すバイオインフォマティクス。技術の発展に伴い、生物学・生命科学の新たな知見が数多く得られることを期待したいです。
記事執筆:吉田拓実(東京大学大学院 農学生命科学研究科 博士課程修了 博士(農学)/ 再考編集室 編集記者 / さいこうファーム 農場長)
<参考文献>
『バイオインフォマティクス:生命情報を科考える』
著書:舘野義男
出版:裳華房(2008年)
(上記すべて参照:2024-4-8)

リケラボ編集部
理系の学生/社会人の方が、ハッピーなキャリアを描(えが)けるように、色々な情報を収集して発信していきます! こんな情報が知りたい!この記事についてもっと深く知りたい!といったリクエストがありましたら、お問い合わせボタンからどしどしご連絡ください!






