化学実験、バイオ実験のノウハウなど、毎日の実験・分析に役立つ情報をお届け。

PCR反応を成功させるために
リケラボ実験レシピシリーズ PCR(3/4)
前回、前々回と、PCRを正しく行うための予備知識をお送りしてきました。
いよいよPCR反応を自分で設計して、実際に行なってみましょう。
ここでは、プルーフリーディング活性をもち、正確性と伸長速度のバランスに優れた「Ex Taqポリメラーゼ(タカラバイオ社)」を例に、「PCR反応液」を調製し、「PCRの反応条件」を設定する際の要点をご紹介します。また、PCR結果を正しく「読む」ための手がかりとなる、「対照実験」の重要性についても概説していきます。
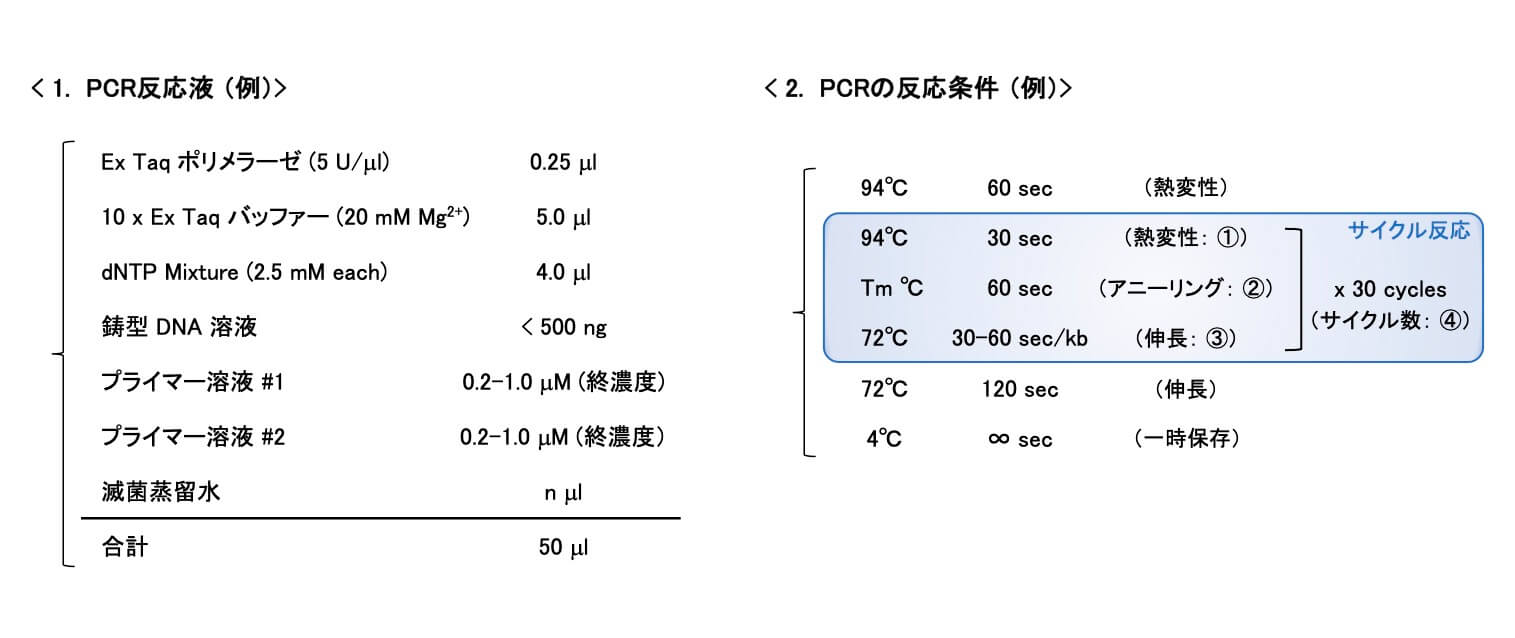
※タップして拡大
1.「PCR反応液」を調製するときのポイント
ポリメラーゼの濃度
- 反応液100μlあたり、酵素2.5Uを目安にする。濃度が高すぎると、非特異的な増幅が生じてしまうので注意!
dNTP(デオキシヌクレオシド三リン酸)の濃度
- 汎用されているTaqポリメラーゼは、dNTP濃度が0.2mMのときに、最大活性を与えるものが多い。
鋳型DNA溶液
- 鋳型DNAが過剰の場合に、非特異的な増幅が生じることがあります。100μlの反応液中に、0.1μg以下の鋳型DNAを含むように調製する。
- 鋳型DNA溶液に含まれるバッファーなどの成分が、PCR反応液の組成を変えてしまうことに注意する。→TEを溶媒とした鋳型DNA溶液は、反応系の1/10量(μl)以下であれば、持ち込みのTEがPCRに大きな悪影響を与えることはない。
→何らかの酵素反応を行った後のDNA溶液を、PCRの鋳型DNAとして用いる場合は、溶液中に存在するMg2+やdNTPなどの物質が、PCR反応に干渉することがあるので注意する!
プライマー濃度
- 最終濃度は0.5μM程度にする。濃度が高すぎると非特異的な増幅が生じてしまうので注意する!
2.「PCRの反応条件」を設定するときのポイント
①熱変性(Denaturing)
- 94°Cで1分程度を目安とする。反応液量が少ないときには、30秒でも十分な熱変性を達成できる場合がある。
- 熱変性の時間が長すぎると、耐熱性ポリメラーゼでも失活してしまうので注意する。
- ゲノムなどの長鎖DNAを鋳型にした場合には、最初の熱変性を2~5分に設定する。
②アニーリング(Annealing)
- アニーリング温度は、一般的にプライマーのTm値またはTm値より数度低い温度で設定する。
- 非特異的な増幅が見られる場合には、アニーリング温度を2°C上げてみる。
- PCR増幅産物が全く見えない場合には、アニーリング温度を2°C下げてみる。[前項PCR2を参照]
③伸長反応(Extension)
- ポリメラーゼの種類にもよるが、通常は72°Cで行う。
- 伸長速度や増幅できる鎖長の上限は、ポリメラーゼの種類によって異なる。Ex Taqポリメラーゼの伸長速度は、1kb/30~60秒程度である。
④サイクル数
- 長時間の加熱を行うと、耐熱性ポリメラーゼでも失活してしまう。酵素が失活すると、それ以降の伸長/増幅が進まなくなる。
- 酵素の失活に関わらず、PCR生成物量がプラトーに達すると、以降のサイクルでは伸長/増幅が起こらない[前項PCR1を参照]。
3.PCRの結果を「正しく」読むポイント コントロール実験の重要性
PCR産物をアガロース電気泳動で確認した際に[アガロース電気泳動の項を参照]、「予想される増幅産物とは異なるバンドが検出された!」、「どのPCRサンプルにも全くバントが検出されない!」といったトラブルに見舞われたことはありませんか?想定外の実験結果を生んだこれらのPCR反応では、一体何が起こっていたのでしょうか?
どんなに熟達した優秀な科学者であっても、この実験結果のみからこの問いに対する「確実に正しい回答」を用意することはできません。しかしながら、「ある実験」を行うと、初学者でも論理的に「正答」にたどり着くことができます。
ここでは、PCRの結果を「正しく」読み解くための手がかり、「対照(コントロール)実験」について考えてみましょう。
3-1.ネガティブコントロール(陰性対照)
PCRでは、他の化学反応では問題視しないほど微量な夾雑DNAが、予期せぬ増幅を生み、目的とは異なるPCR産物を生成してしまうことがあります。
- PCR産物の電気泳動で検出されたバンドが、「鋳型DNAに由来するPCR産物」なのか、「夾雑DNAに由来する増幅産物」なのかを判定するために行う。
→一般的には、「鋳型DNA溶液の代わりに、溶媒(蒸留水やTEなど)を加えた反応液」をネガティブコントロールとして用いる。
→「標的配列を持たないことが確認済み、かつ鋳型DNAと塩基配列・組成・質が同等なDNA試料」を入手できる場合には、これが理想的なネガティブコントロールになる。
3-2.ポジティブコントロール(陽性対照)
PCR産物のアガロース電気泳動で、「全サンプルに、バンドが全く検出されなかった。」場合、考えられる原因は、主に次の2つになります。「反応液中に鋳型が存在しなかったから」もしくは「PCR反応自体に根本的な問題があったから」です。原因がどちらになるのかを明確にし、トラブルシュートをするためには、ポジティブコントロールが必要となります。
ポジティブコントロールは、その目的の違いから、次の2種類に分けられます。
3-2-1.PCR反応のポジティブコントロール
- 特定DNAの存在/非存在を検定する際に行われるコントロール(例:あるウイルスや細菌が試料中に存在するか否かをPCRで検定する場合など)。
- PCR反応後に増幅産物が検出されなかった場合に、「単純な実験ミス」が原因である可能性を検証/否定するために行う。
→「PCR産物が得られることが確認済みのプライマー」と「そのプライマーに対応する鋳型DNA」をペアで用いたPCRをコントロールとして行う。
→このポジティブコントロールでも増幅が見られない場合は、単純な実験ミス(酵素の失活、PCR条件の不良、プライマーの入れ忘れなど)が懸念される。
3-2-2.サンプルのポジティブコントロール
- 3-2-1に合格し、単純な実験ミスは無いと結論付けられたPCR反応系において、増幅産物が検出されないときに行うコントロール実験。
- PCRの反応物として用いた、鋳型DNAの質が十分であるかを検証するために行う。
→「PCR産物が得られることを、別のDNA試料を用いてあらかじめ確認されている別のプライマー」と「そのプライマーの標的配列を確かに持つDNA」のペアを用いて行うPCRが、このポジティブコントロールになる。
→このコントロール実験でPCR産物が検出されれば、用いたDNA溶液が、PCRの鋳型として十分な質のDNAを含むことが明らかになります。コントロール実験は、ひと手間ふた手間かかるうえに、試薬もその分消耗していまうことから、ついつい省略してしまいたくなります。しかしながら、コントロールを省略し、結果の解釈に保証がない状態で実験を続けると、再現性が疑われる実験結果を抱えたまま研究を進める「リスク」が生じます。さらに、この解釈が誤っていた場合には、「時間・貴重なサンプル・労力・資金の浪費」へと繋がります。コントロールを道しるべとして、1歩ずつに確信を持って進んでいくことができるように実験をデザインする必要があります。
次回は、PCRの応用例として、「部位選択的な遺伝子変異の導入法」についてご紹介します!
*監修
パーソルテンプスタッフ株式会社
研究開発事業本部(Chall-edge/チャレッジ)
研修講師(理学博士)
この記事は、理系研究職の方のキャリア支援を行うパーソルテンプスタッフ研究開発事業本部(Chall-edge/チャレッジ)がお届けする、実験ノウハウシリーズです。
過去の記事一覧:実験レシピシリーズ

リケラボ編集部
理系の学生/社会人の方が、ハッピーなキャリアを描(えが)けるように、色々な情報を収集して発信していきます!
こんな情報が知りたい!この記事についてもっと深く知りたい!といったリクエストがありましたら、お問い合わせボタンからどしどしご連絡ください!
求人をお探しの方はこちら

関連記事Recommend
-

細胞の免疫蛍光染色 実践編(2/2)
リケラボ実験レシピシリーズ
-

細胞の免疫蛍光染色 基本編(1/2)
リケラボ実験レシピシリーズ
-

細胞内でRNA干渉を人為的に誘導するために RNA干渉による遺伝子発現の調節(2/2)
リケラボ実験レシピシリーズ
-

RNA干渉(RNA interfering, RNAi)とは? RNA干渉による遺伝子発現の調節(1/2)
リケラボ実験レシピシリーズ
-

免疫沈降法(Immunoprecipitation)による目的タンパク質の濃縮 基本から応用まで
リケラボ実験レシピシリーズ
-

ウェスタンブロットによるタンパク質の発現解析(3/3)ウェスタンブロットによるタンパク質の検出
リケラボ実験レシピシリーズ
-

ウェスタンブロットによるタンパク質の発現解析(2/3)培養細胞抽出液のSDSポリアクリルアミド電気泳動
リケラボ実験レシピシリーズ
-

ウェスタンブロットによるタンパク質の発現解析(1/3)タンパク質実験をはじめるための基礎知識
リケラボ実験レシピシリーズ
-

リポフェクション法による遺伝子導入のプロトコル
リケラボ実験レシピシリーズ トランスフェクション(2/2)
-

遺伝子導入技術 トランスフェクションの基礎知識
リケラボ実験レシピシリーズ トランスフェクション(1/2)